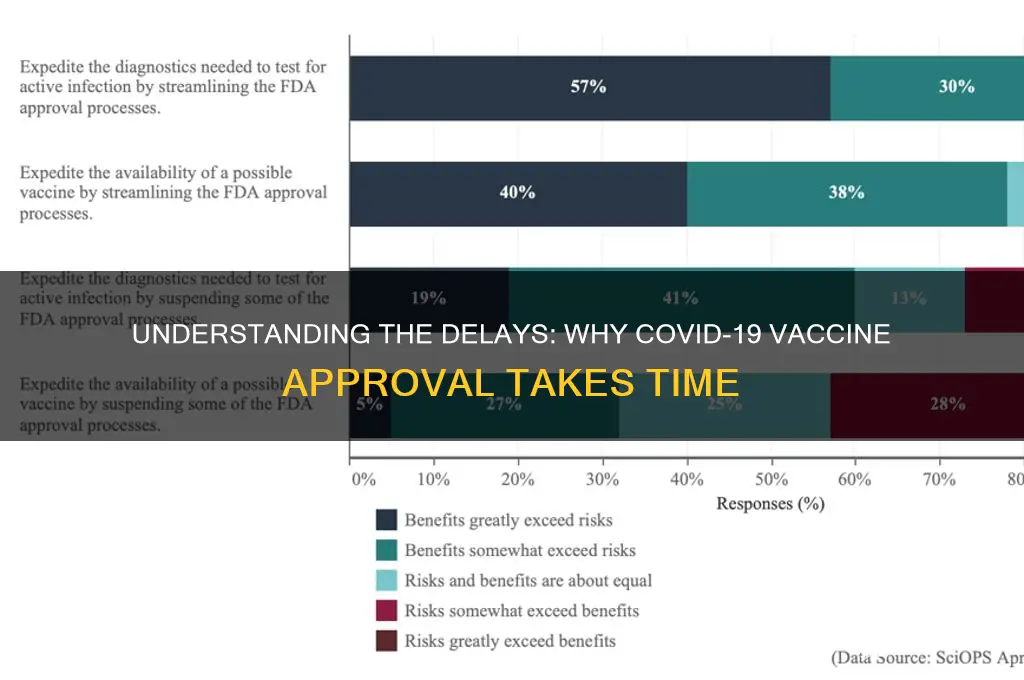
The question of why a vaccine hasn't been approved yet often arises during public health crises, reflecting a mix of scientific rigor, regulatory processes, and public scrutiny. Vaccine development is a complex, multi-stage process that includes preclinical testing, clinical trials, and extensive safety and efficacy evaluations. Regulatory bodies, such as the FDA or EMA, require robust data to ensure the vaccine is both safe and effective before granting approval. Delays can occur due to insufficient trial data, manufacturing challenges, or the need for additional studies to address specific concerns, such as rare side effects or long-term immunity. Additionally, public trust and transparency play a critical role, as rushed approvals can undermine confidence in the vaccine. While the timeline may seem slow, these steps are essential to ensure the vaccine meets stringent standards and protects public health effectively.
Explore related products






What You'll Learn
![]()
Regulatory review process timelines
The regulatory review process for vaccine approval is a meticulous journey, often spanning months or even years. It’s not merely a bureaucratic hurdle but a critical safeguard to ensure safety, efficacy, and quality. For instance, the U.S. Food and Drug Administration (FDA) requires manufacturers to submit detailed data from clinical trials, including information on dosage levels—such as a 30-microgram dose for adults versus a lower 10-microgram dose for children—to assess how the vaccine performs across different age groups. This step-by-step scrutiny is non-negotiable, even in urgent situations like a pandemic.
Consider the phases of regulatory review: preclinical testing, three clinical trial phases, and manufacturing inspections. Each phase has strict timelines. For example, Phase 3 trials typically involve thousands of participants and can last 6–12 months, depending on the disease’s prevalence in the population. After trials, the FDA’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) meets to evaluate data, a process that can take 2–3 months. Even expedited pathways like Emergency Use Authorization (EUA) require at least 2 months of safety data post-vaccination. These timelines aren’t arbitrary delays but deliberate measures to identify rare side effects that smaller, shorter trials might miss.
A common misconception is that regulators are slow or inefficient. In reality, the process is designed to balance speed with rigor. For example, during the COVID-19 pandemic, the FDA reviewed Pfizer’s vaccine application in just 18 days, compared to the usual 6–10 months. However, this acceleration relied on rolling submissions, where data was reviewed as it became available, not after trials concluded. Practical tip: Manufacturers can shorten timelines by initiating rolling submissions early, but this requires flawless coordination and transparency.
Comparatively, regulatory bodies in different countries may have varying timelines due to distinct requirements. The European Medicines Agency (EMA) often takes 1–2 months longer than the FDA for final approval, as it involves additional consultations with EU member states. Meanwhile, countries with less stringent regulations might approve vaccines faster, but at the risk of overlooking critical safety data. For instance, a vaccine approved in one country without robust Phase 3 data might later face global scrutiny if adverse effects emerge.
In conclusion, regulatory review timelines aren’t a bottleneck but a necessary framework to protect public health. Understanding these timelines—from dosage-specific trials to cross-country regulatory differences—helps demystify why vaccine approval takes time. Patience isn’t passive; it’s a commitment to science and safety. Practical takeaway: Stay informed about the specific regulatory milestones for a vaccine, as these indicate progress rather than delay.
Pennsylvania's Vaccine Distribution: Who's Leading the Effort and How?
You may want to see also
Explore related products






![]()
Safety and efficacy data requirements
Vaccine approval hinges on rigorous safety and efficacy data, a process that demands time, precision, and transparency. Regulatory bodies like the FDA and EMA require extensive clinical trial data to ensure a vaccine’s benefits outweigh its risks. For instance, Phase 3 trials typically involve tens of thousands of participants across diverse demographics to assess how the vaccine performs in real-world conditions. These trials must demonstrate not only that the vaccine prevents disease but also that it does so consistently across age groups, including vulnerable populations like the elderly or immunocompromised individuals. Without such comprehensive data, approval cannot proceed.
Consider the specific requirements for efficacy data. A vaccine must achieve a predefined threshold, often around 50% or higher, to be considered effective. For example, the Pfizer-BioNTech COVID-19 vaccine demonstrated 95% efficacy in preventing symptomatic infection in its Phase 3 trial, a benchmark that set a high standard for subsequent vaccines. However, achieving such results requires meticulous trial design, including placebo-controlled groups and long-term follow-up to monitor for waning immunity or rare side effects. If a vaccine fails to meet these efficacy benchmarks or if the data is inconclusive, approval is delayed until further evidence is gathered.
Safety data is equally critical, if not more so, as it directly impacts public trust. Regulatory agencies scrutinize adverse event reports, requiring manufacturers to track everything from mild reactions like soreness at the injection site to severe, life-threatening conditions. For example, the rare cases of thrombosis with thrombocytopenia syndrome (TTS) linked to the Johnson & Johnson vaccine led to temporary pauses in its rollout while investigators assessed the risk. Such incidents underscore the importance of robust safety monitoring systems, including post-authorization surveillance, to detect and address issues that may not appear in initial trials.
Practical considerations also play a role in meeting safety and efficacy requirements. Dosage regimens, for instance, must be optimized to balance protection and side effects. The Moderna COVID-19 vaccine initially used a 100-microgram dose for adults but later explored half-dose regimens for booster shots to minimize adverse reactions while maintaining efficacy. Similarly, age-specific trials are often required to ensure safety in children, whose immune systems differ significantly from adults. For example, the Pfizer vaccine was approved for adolescents aged 12–15 only after separate trials confirmed its safety and efficacy in this age group.
In conclusion, the delay in vaccine approval often stems from the meticulous process of gathering and analyzing safety and efficacy data. This process is not merely bureaucratic but essential for ensuring public health. From establishing clear efficacy thresholds to monitoring rare adverse events and tailoring dosages for specific populations, every step is designed to protect individuals and communities. While the wait can be frustrating, it is a testament to the commitment to scientific rigor and ethical standards that underpin vaccine development.
Selling Puppies Without Vaccinations: Legal or Unlawful Practice?
You may want to see also
Explore related products






![]()
Manufacturing quality control challenges
Ensuring consistent quality across millions of vaccine doses is a logistical nightmare, particularly when scaling production to meet global demand. Each manufacturing site must adhere to stringent Good Manufacturing Practices (GMP), but even minor deviations in temperature, humidity, or equipment calibration can compromise the final product. For instance, mRNA vaccines like Pfizer-BioNTech’s require storage at -70°C, and any fluctuation during production or transportation can degrade the lipid nanoparticles encapsulating the mRNA, rendering doses ineffective. Regulatory agencies scrutinize every step, from raw material sourcing to final packaging, often requiring multiple inspections before granting approval.
Consider the complexity of scaling up production while maintaining precision. A single batch of a viral vector vaccine, such as AstraZeneca’s, involves culturing cells in bioreactors, introducing the vector, and purifying the product—a process that can take weeks. If any step is rushed or improperly executed, contaminants like endotoxins or residual host cell proteins may remain, triggering adverse reactions in recipients. Manufacturers must also ensure uniformity in dosage, typically measured in micrograms (e.g., 30 µg for Moderna’s vaccine). Even a slight variation can affect efficacy or safety, particularly in vulnerable populations like the elderly or immunocompromised.
One practical challenge lies in validating manufacturing processes across different facilities. When a vaccine is produced in multiple countries, each site must replicate the exact conditions of the original trial batches. For example, differences in water quality or filtration systems can introduce variability. Regulators often require comparative studies to prove that doses produced in, say, India or Brazil are indistinguishable from those made in the U.S. or Europe. This process, known as comparability testing, can delay approval by months, as agencies demand robust data to ensure equivalence.
To mitigate these risks, manufacturers employ rigorous in-process controls, such as real-time monitoring of pH levels, oxygen concentration, and cell viability. However, even with advanced technology, human error remains a wildcard. Training staff to follow protocols precisely is critical, especially when handling sensitive materials like live attenuated viruses. For instance, a technician’s failure to sterilize equipment properly could introduce microbial contamination, necessitating an entire batch’s destruction. Such incidents not only delay production but also erode public trust in the vaccine’s safety.
Ultimately, manufacturing quality control challenges are a bottleneck in vaccine approval, but they are non-negotiable. Every dose must meet the same exacting standards, regardless of where or when it’s produced. While these hurdles may slow the process, they are essential to ensuring that vaccines are both safe and effective. For those awaiting approval, understanding these complexities underscores why patience is as vital as the vaccine itself.
Oregon Vaccine Eligibility: Who Qualifies for COVID-19 Shots Now?
You may want to see also
Explore related products






![]()
Political and public pressure impact
Political pressure can significantly delay vaccine approval, often overshadowing scientific rigor. Governments, eager to demonstrate control over public health crises, may push regulatory bodies to expedite reviews. For instance, during the COVID-19 pandemic, leaders in multiple countries publicly set ambitious timelines for vaccine rollout, implicitly pressuring health agencies to accelerate processes. While this urgency can motivate efficiency, it risks compromising safety checks, such as extended phase III trials that typically involve tens of thousands of participants over 12–18 months to ensure efficacy and identify rare side effects. Regulatory bodies like the FDA or EMA must balance political demands with their mandate to protect public health, creating a tension that can prolong approval timelines as they navigate scrutiny from both policymakers and the public.
Public pressure, fueled by misinformation and fear, further complicates the approval process. Social media campaigns, often amplified by high-profile figures, can create unrealistic expectations or sow distrust in vaccines under development. For example, during the H1N1 pandemic, public skepticism about vaccine safety led to lower uptake rates, prompting regulators to spend additional time addressing concerns through transparency initiatives. Similarly, in the case of COVID-19, rumors about microchips or fertility issues forced health agencies to allocate resources to debunk myths rather than focus solely on scientific evaluation. This diversion of effort can delay approvals, as agencies must engage in extensive public education campaigns to rebuild trust and ensure informed consent.
A comparative analysis of vaccine approval timelines reveals how political and public pressure differ across regions. In the U.S., the FDA’s Emergency Use Authorization (EUA) pathway allowed COVID-19 vaccines to be approved within months, but this speed was met with public skepticism in some quarters, requiring additional post-approval monitoring. In contrast, the European Medicines Agency (EMA) took a more deliberate approach, opting for full approval rather than expedited pathways, which prolonged the process but may have bolstered public confidence. Such regional differences highlight how political priorities and public sentiment shape regulatory strategies, with each approach carrying trade-offs between speed and trust.
To mitigate the impact of political and public pressure, regulators must adopt transparent communication strategies. For instance, publishing detailed trial data, including dosage information (e.g., 30 µg of mRNA in Pfizer’s vaccine) and age-specific efficacy rates (e.g., 95% for adults aged 16–55), can help build credibility. Additionally, engaging community leaders to address local concerns and providing clear instructions on vaccine administration (e.g., two doses spaced 21–28 days apart) can reduce misinformation. By prioritizing openness and inclusivity, regulators can navigate external pressures while maintaining scientific integrity, ensuring vaccines are both safe and widely accepted.
Maryland's Vaccination Progress: How Many Adults Have Received COVID-19 Shots?
You may want to see also
Explore related products






![]()
Global supply chain constraints
The global rollout of vaccines often hinges on supply chain logistics, a complex web of manufacturing, transportation, and distribution that can delay approvals and availability. Consider the COVID-19 vaccine: while clinical trials progressed rapidly, scaling production to billions of doses exposed vulnerabilities in this system. Key raw materials, like lipid nanoparticles essential for mRNA vaccines, faced shortages due to limited global suppliers. A single manufacturing facility disruption, as seen in early 2021 with the AstraZeneca plant in Baltimore, can halt production lines, delaying regulatory submissions and approvals in regions reliant on those doses.
Analyzing the supply chain reveals a domino effect of constraints. Cold chain requirements for vaccines like Pfizer-BioNTech’s, which must be stored at -70°C, demand specialized equipment and infrastructure. Many low-income countries lack ultra-low temperature freezers, leading to distribution bottlenecks and wastage. Even in developed nations, last-mile delivery challenges arise, such as ensuring rural areas receive doses within the 30-minute window post-thawing. These logistical hurdles force regulatory bodies to scrutinize distribution plans, slowing approvals until viable solutions are presented.
To address these constraints, a multi-faceted approach is necessary. First, diversify manufacturing hubs to reduce dependency on single sources. For instance, India’s Serum Institute played a critical role in producing AstraZeneca doses, but its capacity was stretched thin. Second, invest in local cold chain infrastructure, such as solar-powered refrigerators, to ensure vaccine stability in remote regions. Third, streamline regulatory harmonization across countries to avoid redundant approval processes that delay distribution. Practical steps like these can mitigate supply chain bottlenecks, accelerating vaccine availability globally.
Comparatively, the H1N1 vaccine in 2009 faced similar but less severe supply chain issues, as its production relied on traditional egg-based methods with established infrastructure. In contrast, the novel mRNA technology for COVID-19 vaccines required new materials and processes, amplifying constraints. This highlights the need for proactive supply chain planning in vaccine development, treating logistics as integral to approval timelines, not an afterthought. Without such foresight, even the most effective vaccines risk languishing in regulatory limbo.
NHS App: Your Vaccine Status at Your Fingertips
You may want to see also
Frequently asked questions
Vaccine development involves multiple phases of clinical trials, regulatory reviews, and safety assessments, which can take months to years to ensure efficacy and safety before approval.
Regulatory agencies in different countries have varying approval processes and timelines. Some may prioritize emergency use authorizations, while others wait for more comprehensive data before granting full approval.
Emergency use authorization (EUA) is a temporary measure during public health crises. Full approval requires additional long-term data on safety and efficacy, which takes more time to collect and review.
While high efficacy is promising, regulators need to thoroughly evaluate all trial data, including rare side effects, long-term outcomes, and manufacturing quality, to ensure the vaccine meets safety standards.
Vaccines distributed under emergency use or in clinical trials are closely monitored, but full approval requires a more extensive review process to ensure it’s safe and effective for widespread, long-term use.