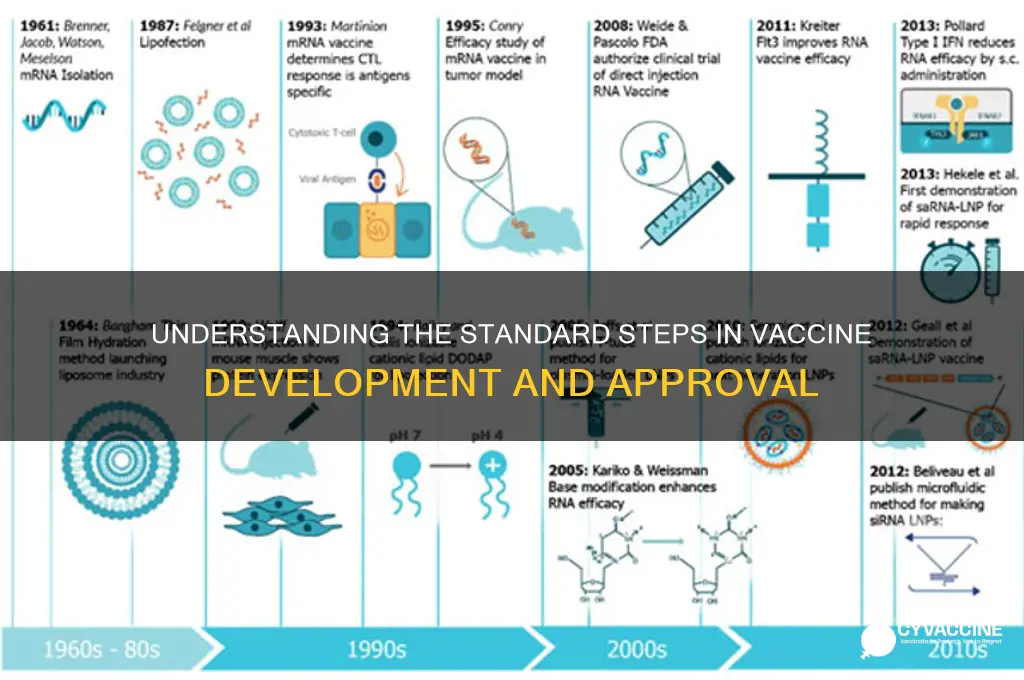
Vaccine development is a rigorous, multi-stage process designed to ensure safety, efficacy, and quality before a vaccine is approved for public use. It typically begins with exploratory research, where scientists identify antigens that can stimulate the immune system to protect against a specific disease. This is followed by pre-clinical testing, where potential vaccines are tested in lab settings and animal models to assess safety and immunogenicity. If successful, the vaccine advances to clinical trials, which are conducted in three phases: Phase I evaluates safety and dosage in a small group of healthy volunteers, Phase II expands testing to assess efficacy and side effects in a larger population, and Phase III involves thousands of participants to confirm effectiveness and monitor rare side effects. After clinical trials, regulatory agencies review the data to approve the vaccine, followed by large-scale manufacturing and distribution. Post-approval, ongoing surveillance (Phase IV) ensures long-term safety and efficacy in the general population. This entire process, from initial research to market availability, can take 10–15 years, though expedited timelines, such as those seen during the COVID-19 pandemic, are possible under emergency circumstances.
| Characteristics | Values |
|---|---|
| Exploratory Stage | 2-5 years; involves identifying antigens and potential vaccine candidates. |
| Pre-Clinical Stage | 1-2 years; testing in animals to assess safety and efficacy. |
| Clinical Development (Phase 1) | 1-2 years; small-scale human trials (20-100 volunteers) to test safety. |
| Clinical Development (Phase 2) | 2-3 years; larger trials (100-300 volunteers) to assess immunogenicity. |
| Clinical Development (Phase 3) | 3-5 years; large-scale trials (thousands to tens of thousands) to confirm efficacy and safety. |
| Regulatory Review and Approval | 6 months to 2 years; submission and evaluation by regulatory agencies (e.g., FDA, EMA). |
| Manufacturing and Quality Control | Ongoing; scaling up production and ensuring consistent quality. |
| Phase 4 (Post-Market Surveillance) | Ongoing; monitoring vaccine safety and efficacy in the general population. |
| Total Timeline (Traditional Process) | 10-15 years on average. |
| Accelerated Timeline (e.g., COVID-19) | 1-2 years; achieved through parallel processing, funding, and global collaboration. |
| Cost | $500 million to $2 billion per vaccine, including failures. |
| Success Rate | Only 6% of vaccine candidates make it from pre-clinical to approval. |
| Key Technologies | mRNA, viral vectors, protein subunits, whole-virus (attenuated/inactivated). |
| Regulatory Agencies | FDA (U.S.), EMA (Europe), WHO (global), etc. |
| Funding Sources | Government grants, private investments, international organizations (e.g., CEPI, Gavi). |
Explore related products






What You'll Learn
- Pre-clinical Testing: Lab and animal studies to assess safety and efficacy before human trials
- Clinical Trials: Phased human testing to evaluate safety, dosage, and immune response
- Regulatory Review: Submission to health authorities for approval based on trial data
- Manufacturing Scale-Up: Production of vaccine doses under strict quality control standards
- Distribution & Monitoring: Global rollout and post-vaccination surveillance for side effects
![]()
Pre-clinical Testing: Lab and animal studies to assess safety and efficacy before human trials
Before any vaccine candidate reaches human trials, it undergoes rigorous pre-clinical testing—a critical phase that bridges the gap between theoretical science and practical application. This stage involves meticulous laboratory and animal studies designed to evaluate both the safety and efficacy of the vaccine. The goal is to identify potential risks, optimize dosage, and ensure the vaccine triggers the desired immune response before it is administered to humans. Without this step, the leap to clinical trials would be reckless, jeopardizing both scientific integrity and public trust.
In the lab, researchers begin by testing the vaccine candidate on cell cultures to assess its ability to stimulate an immune response. This initial step often involves exposing cells to the vaccine antigen—whether it’s a weakened virus, a protein subunit, or a genetic material like mRNA. For example, in the development of the COVID-19 mRNA vaccines, lab studies confirmed that the mRNA sequences encoding the SARS-CoV-2 spike protein could effectively prompt cells to produce the target antigen. Dosage is a key focus here; researchers experiment with varying concentrations (e.g., 10 µg, 25 µg, or 50 µg) to determine the minimum effective dose that maximizes immune response while minimizing side effects. This phase also includes toxicity tests to ensure the vaccine components are safe at the intended dosage levels.
Animal studies represent the next critical step in pre-clinical testing. These trials are conducted on species that closely mimic human immune responses, such as mice, ferrets, or non-human primates. For instance, influenza vaccine candidates are often tested in ferrets due to their susceptibility to the virus and similar respiratory physiology to humans. Animals are divided into control and test groups, with the latter receiving the vaccine candidate. Researchers then expose both groups to the pathogen to compare infection rates, disease severity, and immune responses. A successful outcome would show that vaccinated animals have significantly lower viral loads or milder symptoms compared to the control group. These studies also monitor for adverse reactions, such as allergic responses or organ toxicity, which could indicate the need to refine the vaccine formulation.
One of the most persuasive arguments for the necessity of pre-clinical testing lies in its ability to predict potential failures before human trials. For example, during the development of a cytomegalovirus (CMV) vaccine, pre-clinical studies in rhesus macaques revealed that the vaccine, while safe, failed to induce a robust enough immune response to prevent infection. This finding allowed researchers to halt the project before investing in costly and potentially futile clinical trials. Such examples underscore the value of pre-clinical testing as a safeguard against wasted resources and, more importantly, harm to human participants.
In conclusion, pre-clinical testing is a non-negotiable cornerstone of vaccine development. It combines laboratory precision with animal model insights to ensure that only the safest and most promising candidates advance to human trials. By systematically evaluating dosage, efficacy, and safety, this phase not only protects future trial participants but also accelerates the path to a viable vaccine. Skipping or rushing this step would undermine the entire process, risking both scientific credibility and public health.
IJCE Journal: Exploring Political Issues and Vaccine Policies
You may want to see also
Explore related products


$246.55 $84.99




![]()
Clinical Trials: Phased human testing to evaluate safety, dosage, and immune response
Clinical trials are the backbone of vaccine development, ensuring that new immunizations are both safe and effective before they reach the public. These trials are meticulously phased, each stage designed to answer specific questions about the vaccine’s safety, optimal dosage, and ability to elicit an immune response. Phase 1 trials typically involve a small group of healthy volunteers, often 20 to 100 individuals, to assess safety and identify potential side effects. Participants are closely monitored, and dosages are carefully escalated to determine the highest amount that remains safe and tolerable. For example, in the development of the COVID-19 vaccines, Phase 1 trials tested doses ranging from 10 to 200 micrograms to find the sweet spot that balanced efficacy and side effects.
Once safety is established, Phase 2 trials expand to include several hundred participants, often targeting specific age groups or populations at higher risk. This phase refines the dosage and evaluates the vaccine’s ability to generate an immune response. For instance, in pediatric vaccine trials, children aged 5–12 might receive a lower dose than adults to account for differences in body weight and immune system maturity. Researchers also analyze biomarkers, such as antibody levels, to gauge how well the vaccine primes the immune system. Practical tips for participants include maintaining a symptom diary and attending all scheduled follow-up visits to ensure accurate data collection.
Phase 3 trials are the largest and most critical, involving thousands to tens of thousands of participants across diverse demographics and geographic locations. This phase tests the vaccine’s efficacy in preventing disease in real-world conditions. Placebos are often used to create a control group, and participants are randomly assigned to ensure unbiased results. For example, the Pfizer-BioNTech COVID-19 vaccine’s Phase 3 trial included over 43,000 participants, demonstrating 95% efficacy in preventing symptomatic infection. Ethical considerations, such as ensuring informed consent and providing access to the vaccine after the trial, are paramount in this phase.
Throughout these phases, transparency and rigor are essential. Regulatory agencies like the FDA and WHO scrutinize trial data to ensure vaccines meet stringent safety and efficacy standards. For instance, emergency use authorizations during the COVID-19 pandemic required at least two months of safety data following vaccination, highlighting the balance between urgency and caution. Clinical trials are not just a bureaucratic hurdle but a scientific imperative, ensuring that vaccines protect without harm. Their phased approach allows for incremental learning, reducing risks while advancing medical breakthroughs.
Understanding the Chickenpox Vaccine: Is It a Two-Part Series?
You may want to see also
Explore related products






![]()
Regulatory Review: Submission to health authorities for approval based on trial data
Regulatory review is the critical juncture where years of vaccine research and clinical trials meet the scrutiny of health authorities. This phase is not merely a formality but a rigorous evaluation process designed to ensure safety, efficacy, and quality before a vaccine reaches the public. Manufacturers must compile and submit a comprehensive dossier, often thousands of pages long, detailing every aspect of the vaccine’s development, from preclinical studies to Phase III trial results. For instance, the Pfizer-BioNTech COVID-19 vaccine submission included data from over 43,000 trial participants, demonstrating 95% efficacy in preventing symptomatic infection. This submission is the culmination of meticulous documentation, ensuring that every question a regulator might ask is preemptively addressed.
The regulatory review process varies by country but typically involves a multi-step evaluation. In the United States, the FDA scrutinizes the data through its Center for Biologics Evaluation and Research (CBER), assessing not only clinical outcomes but also manufacturing consistency and facility inspections. Similarly, the European Medicines Agency (EMA) employs a rolling review process, allowing regulators to assess data as it becomes available, expediting approval without compromising standards. A key focus is on the risk-benefit analysis: does the vaccine’s protective effect outweigh potential side effects? For example, the Moderna vaccine’s approval included a detailed examination of rare cases of myocarditis, particularly in young males, leading to specific dosage recommendations (100 µg for adults, 50 µg for adolescents).
One of the most challenging aspects of regulatory review is balancing speed with thoroughness, especially during public health emergencies. Emergency Use Authorizations (EUAs) provide a mechanism for accelerated approval, as seen during the COVID-19 pandemic, but even these require robust data. Regulators must ensure that shortcuts in timelines do not compromise safety. For instance, the AstraZeneca vaccine’s approval in the UK included a stipulation for ongoing monitoring of rare blood clotting events, illustrating how post-authorization surveillance becomes an integral part of the regulatory framework. This adaptive approach ensures that any emerging risks are swiftly addressed.
Practical tips for manufacturers navigating this phase include early engagement with regulators, such as through the FDA’s Breakthrough Therapy designation, which provides expedited review for vaccines targeting serious conditions. Additionally, transparency in reporting adverse events, even if rare, builds trust and facilitates smoother approvals. For example, Johnson & Johnson’s submission included detailed data on vaccine-induced immune thrombotic thrombocytopenia (VITT), leading to targeted warnings for specific age groups (e.g., women under 50). Such specificity ensures that vaccines are deployed safely and effectively, tailored to the needs of diverse populations.
In conclusion, regulatory review is a cornerstone of vaccine development, bridging scientific innovation with public health protection. It demands precision, transparency, and adaptability from both manufacturers and regulators. By adhering to stringent standards while remaining responsive to urgent needs, this phase ensures that vaccines not only meet theoretical benchmarks but also deliver real-world impact. Whether through standard approvals or emergency pathways, the goal remains the same: to provide safe, effective vaccines that save lives.
Spouse Vaccination: Essential Workers' Eligibility
You may want to see also
Explore related products


$48.53 $96.95




![]()
Manufacturing Scale-Up: Production of vaccine doses under strict quality control standards
The transition from laboratory-scale production to mass manufacturing is a critical phase in vaccine development, where the focus shifts from creating a viable product to ensuring its availability for widespread use. This scale-up process is a complex endeavor, requiring meticulous planning and execution to maintain the vaccine's safety, efficacy, and consistency. Imagine a recipe that works perfectly for a small family dinner but needs to be adapted to feed an entire community without compromising taste or quality—this is the challenge manufacturers face.
The Art of Scaling Up:
Manufacturing scale-up involves a series of strategic steps. Firstly, the production process is optimized to increase yield while maintaining product quality. This might include modifying the growth conditions for cells or microorganisms used in vaccine production, or adjusting the formulation to ensure stability at a larger scale. For instance, the influenza vaccine production often utilizes egg-based or cell-based methods, each requiring specific conditions for optimal virus growth and harvest. The scale-up process must consider these unique requirements to ensure the vaccine's effectiveness.
Quality Control: A Non-Negotiable Priority
As production scales up, quality control becomes even more stringent. Every batch of the vaccine must meet predefined standards, ensuring consistency and safety. This involves rigorous testing at various stages of production. For example, each batch of the measles, mumps, and rubella (MMR) vaccine undergoes tests to confirm the potency of each viral component, ensuring it falls within the specified range of 10^3.0^ to 10^5.0^ TCID50 (Tissue Culture Infectious Dose) per dose. Any deviation from these standards could render the vaccine ineffective or, worse, harmful.
Practical Considerations:
- Facility Design: Manufacturing facilities are designed with scale-up in mind, incorporating flexible production suites that can accommodate varying batch sizes. These suites are often equipped with single-use technologies, reducing the risk of cross-contamination and simplifying the transition between different vaccine productions.
- Supply Chain Management: Scaling up production also means managing a complex supply chain. Manufacturers must ensure a steady supply of raw materials, from cell culture media to vial stoppers, to meet the increased demand. This requires careful planning and collaboration with suppliers to avoid shortages.
- Regulatory Compliance: Throughout the scale-up process, manufacturers must adhere to strict regulatory guidelines. These regulations dictate everything from the qualification of equipment to the validation of cleaning procedures, ensuring that every step meets the required standards.
A Delicate Balance:
The scale-up process is a delicate balance between increasing production capacity and maintaining the integrity of the vaccine. It requires a deep understanding of the product, meticulous planning, and a commitment to quality. Manufacturers must navigate this phase with precision, ensuring that every dose produced is safe, effective, and ready to contribute to public health. This stage is where the promise of a vaccine becomes a tangible reality, ready to be distributed and administered to those in need.
Understanding Serious Vaccine Reactions: Symptoms, Risks, and When to Seek Help
You may want to see also
Explore related products






![]()
Distribution & Monitoring: Global rollout and post-vaccination surveillance for side effects
Once a vaccine is approved, the logistical challenge of distributing it globally becomes paramount. This phase requires meticulous planning to ensure equitable access, especially in low-resource settings. For instance, the COVID-19 vaccine rollout highlighted the need for cold chain infrastructure, as some vaccines (like Pfizer-BioNTech) require ultra-low temperatures (-70°C) for storage. In contrast, others (like AstraZeneca) are more stable at standard refrigeration temperatures (2–8°C), making them more suitable for regions with limited resources. Distribution strategies must account for these differences, often involving partnerships with international organizations like Gavi and UNICEF to reach remote areas.
Post-vaccination surveillance is equally critical to monitor safety and efficacy in real-world populations. Passive surveillance systems, such as the Vaccine Adverse Event Reporting System (VAERS) in the U.S., rely on healthcare providers and individuals to report side effects. However, these systems can underreport due to reliance on voluntary participation. Active surveillance, like the CDC’s V-safe program, uses smartphone-based tools to collect data directly from vaccine recipients, offering more comprehensive insights. For example, V-safe identified rare cases of myocarditis in young males after mRNA COVID-19 vaccines, prompting dosage adjustments for certain age groups (e.g., lower doses for children aged 5–11).
A key challenge in monitoring is distinguishing between vaccine-related side effects and coincidental health events. For instance, a headache or fever within 48 hours of vaccination is common and expected, while severe reactions like anaphylaxis are rare (occurring in approximately 2–5 cases per million doses). Pharmacovigilance programs use statistical methods to detect signals of potential safety issues, such as the proportional reporting ratio (PRR), which compares observed and expected rates of adverse events. When a signal is detected, regulatory bodies may issue guidelines, such as the temporary pause of the Johnson & Johnson vaccine in 2021 to investigate rare blood clots.
Global rollout also demands cultural sensitivity and community engagement to address vaccine hesitancy. In some regions, misinformation about side effects has fueled skepticism, underscoring the need for transparent communication. For example, in rural India, local health workers used door-to-door campaigns to educate communities about vaccine safety, emphasizing that mild side effects like arm soreness are normal and not indicative of harm. Similarly, in Africa, leveraging trusted leaders and radio broadcasts helped dispel myths and encourage uptake.
In conclusion, successful distribution and monitoring hinge on adaptability, collaboration, and transparency. From tailoring logistics to local conditions to employing advanced surveillance tools, every step must prioritize public trust and safety. As vaccines continue to evolve, so too must the systems designed to deliver and safeguard them, ensuring that the benefits of immunization reach every corner of the globe.
Understanding NY's Vaccine Exemption Law: Implications and Key Changes Explained
You may want to see also
Frequently asked questions
The process begins with exploratory research to identify antigens (substances that trigger an immune response) and potential vaccine candidates. This phase involves laboratory studies and often takes 2–5 years.
After preclinical testing in animals, vaccines undergo three phases of clinical trials in humans. Phase 1 tests safety and dosage, Phase 2 evaluates immune response and side effects, and Phase 3 assesses efficacy and safety in a larger population.
Traditionally, vaccine development takes 10–15 years, but timelines can be accelerated during emergencies, as seen with COVID-19 vaccines, through parallel processing and increased funding.
Regulatory agencies like the FDA (U.S.) or EMA (Europe) review clinical trial data to ensure the vaccine is safe and effective. They also oversee manufacturing practices before granting approval or emergency use authorization.
Post-approval, vaccines undergo Phase 4 monitoring (pharmacovigilance) to detect rare side effects or long-term impacts. This ensures ongoing safety and effectiveness in the general population.











![[Practical Psychopharmacology]: [Translating Findings From Evidence-Based Trials] into [Real-World Clinical Practice] (New Edition) - 2021, Paperback](https://m.media-amazon.com/images/I/51ZF+z0l4tL._AC_UL320_.jpg)